Introduction
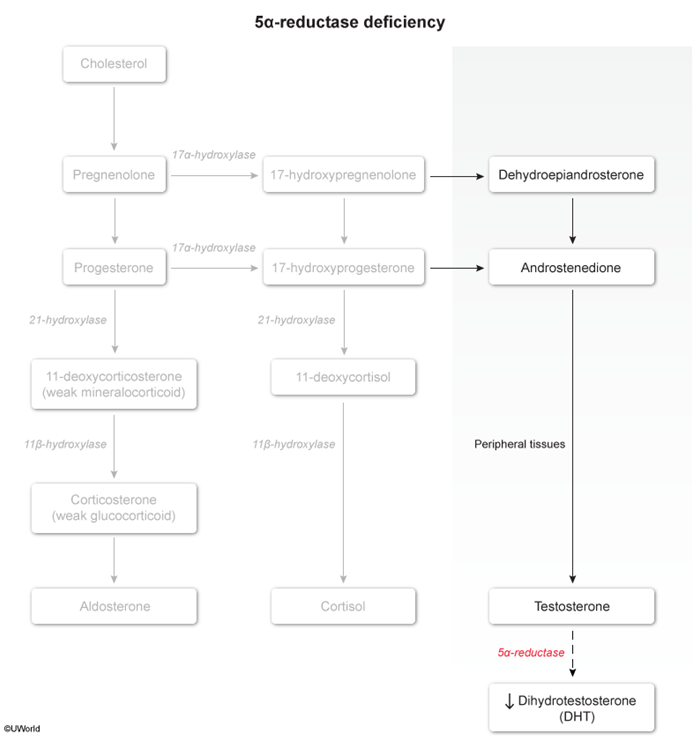
5-alpha reductase deficiency is a rare, autosomal recessive disorder of sex development impairing the conversion of testosterone to dihydrotestosterone (DHT
Pathogenesis
There are 2 types of 5-alpha reductase enzyme that convert testosterone to DHT: type 1 is present in postpubescent skin and type 2 is predominantly found in the genitals. 5-alpha reductase deficiency is caused exclusively by a defective type 2 enzyme. The lack of DHT leads to abnormal sexual development, beginning in utero:
Development of internal genitalia
- In utero, the gonads of genotypic males (46,XY) differentiate into the testes under the control of the sex-determining region of the Y chromosome (SRY gene ( Testicular Leydig cells produce testosterone, which binds to androgen receptors on the wolffian duct, causing proliferation and development into internal male genital ducts (eg, epididymis, vas deferens). This process occurs normally in patients with 5-alpha reductase deficiency because the enzyme is not required for this process.
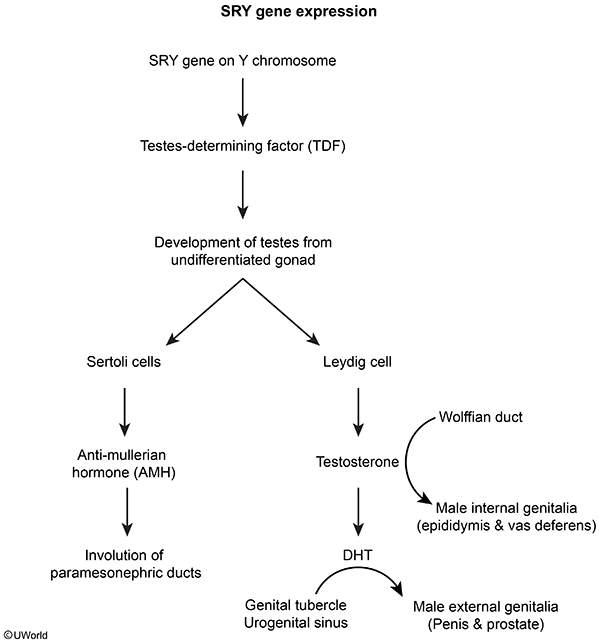 figure 2
figure 2
- Antimüllerian hormone (AMH), produced by testicular Sertoli cells, causes regression of the müllerian structures; therefore, patients with 5-alpha reductase deficiency lack internal female genitalia (eg, fallopian tubes, uterus, proximal vagina).
Development of external genitalia
- Some of the testosterone produced by the testes is also converted to DHT by the enzyme 5-alpha reductase.
- DHT binds androgen receptors on the genital tubercle to promote differentiation into external male genitalia (eg, penis). Because DHT is absent in males with 5-alpha reductase deficiency, the genital tubercle differentiates into female external genitalia (lower two-thirds vagina [ie, blind pouch], vulva). In addition, because DHT mediates testicular descent, the testes are undescended (cryptorchid).
Testosterone levels are within the normal male range due to intact negative feedback within the hypothalamic-pituitary-gonadal axis. However, at puberty, physiologic increases in testosterone lead to virilization of phenotypically female patients through the action of 5-alpha reductase type 1; this can manifest as nodulocystic acne and clitoromegaly (ie, clitoris protruding from the clitoral hood). Additional features of virilization include voice deepening and increased muscle mass. Because testosterone levels are within the normal male range for puberty, patients develop additional, normal secondary sexual characteristics (eg, axillary and pubic hair). However, patients have no breast development because testosterone binds to the breast androgen receptor and inhibits breast tissue proliferation.
Clinical presentation
Clinical features vary based on the timing of clinical presentation.
At birth:
- Undervirilization leads to predominantly female external genitalia; however, features may range from mild clitoromegaly to an isolated microphallus
- +/- bilateral labial masses (cryptorchid testis)
Commonly, patients who are assigned female gender at birth present in adolescence with primary amenorrhea (ie, absence of menarche) or with virilization (eg, clitoromegaly) and the following features:
- No breast development (due to testosterone-inhibited breast proliferation)
- Blind-ending vaginal pouch, absent müllerian structures (eg, uterus, fallopian tubes)
- Normal pubic hair and axillary development
- Virilization characterized by clitoromegaly, increased muscle mass, male pattern hair development, nodulocystic acne
Diagnosis
The diagnosis may occur during the newborn period if ambiguous genitalia (eg, clitoromegaly or microphallus) are present.
However, classically, the diagnosis is delayed until adolescence in those assigned female gender at birth who then present with either primary amenorrhea or rapid virilization. The diagnosis is suspected based on clinical findings and is confirmed through laboratory and imaging findings that include:
Laboratory evaluation
- Elevated testosterone-to-DHT ratio
- Confirmatory genetic testing with identification of the mutated gene that encodes for 5-alpha reductase (SRD5A2)
Imaging
- Ultrasonography to evaluate internal genitalia, reveal absent müllerian structures (eg, uterus, Fallopian tubes), and identify undescended testes
- MRI to visualize detailed anatomy of the pelvic structures in the planning of surgery
Differential diagnosis
The differential diagnosis for 5-alpha reductase deficiency includes other differences of sex development
that present with a female phenotype, primary amenorrhea, and abnormal pubertal development:- Androgen insensitivity syndrome: 46, XY; presents with female external genitalia, primary amenorrhea, and absent müllerian structures (ie, blind-ending vagina); however, these patients have breast development without axillary or pubic hair (due to inability of testosterone to bind nonfunctioning androgen receptors). In addition, the testosterone-to-DHT ratio is normal.
- Müllerian agenesis (ie, Mayer-Rokitansky-Küster-Hauser syndrome): 46,XX; presents with female external genitalia, primary amenorrhea, a blind vaginal pouch, and absent or rudimentary uterus. The ovaries are normal and functioning; therefore, patients have breast development. However, testosterone levels are within the normal female range and axillary/pubic hair is present.
- Female gonadal dysgenesis (eg, Turner syndrome [45,X]) presents with female external genitalia and primary amenorrhea; however, patients have female internal genitalia (uterus, fallopian tubes), streak ovaries, minimal breast development (due to deficient estrogen production), and normal axillary/pubic hair.
- Classic congenital adrenal hyperplasia: 46,XX; presents with ambiguous genitalia due to 21-hydroxylase deficiency, which leads to the loss of mineralocorticoids and glucocorticoids and shunts substrates toward androgen production. However, this also presents with salt-wasting crisis (ie, hypotension, hyponatremia, hyperkalemia, hypoglycemia) in the neonatal period.
Management
Management is individualized based on the degree of genital ambiguity and gender identity. The mainstay of treatment is psychosocial support for individuals and their families to address issues of gender identity. Hormonal therapy can be provided based on gender identity and may require estrogen to maintain bone health (in females) or testosterone supplementation to enhance virilization (in males). Corrective surgery may also be an option for ambiguous genitalia and/or undescended testes.
Prognosis
The overall prognosis of patients with 5-alpha reductase deficiency is good; however, those with unremoved cryptorchid testes are at increased risk of testicular cancer.
Summary
5-alpha reductase deficiency
is a rare genetic disorder affecting male sexual differentiation due to a lack of DHT. Patients have a 46,XY genotype but have a female phenotype (ie, external female genitalia) at birth and experience virilization (eg, clitoromegaly, increased muscle mass) at puberty.